Autoinflammatorischen Erkrankungen
Die autoinflammatorischen Erkrankungen haben die Gemeinsamkeit, dass Fieberschübe in unterschiedlicher Häufigkeit auftreten, meist in Verbindung mit Hautveränderungen und Gelenkentzündungen. Die Erstmanifestation kann sowohl im Kindesalter, als auch im Erwachsenenalter erfolgen.
Ursächlich bestehen meist eine genetisch bedingte Störungen im sogenannten „Inflammasom“, die eine vermehrte Produktion des Zytokins (Botenstoffs) IL-1ß zur Folge haben.
Familiäres Mittelmeerfieber (FMF)
Kommt am häufigsten bei Menschen aus dem Mittelmeerraum vor, in Deutschland also bei Mitbürgern türkischer Herkunft. Ursache ist die Mutation eines Gens. Deutlich wird die Krankheit häufig vor dem 20. Lebensjahr. Die Symptome treten in Schüben auf (ein Schub dauert maximal zwei Tage).
{kind=link}
Hyper-IgD-Syndrom
Hier liegt eine Mutation im MVK (Mevalonatkinase) Gen zugrunde - der Erkrankungsbeginn ligt vor dem 1. Lebensjahr. Die Patienten haben Arthralgien oder eine Oligoarthritis, einen Hautausschlag im Schub, eine zervikale Lymphadenopathie und Fieber. Die Schubdauer liegt bei 4 - 6 Tagen.
Cryopyrin assoziierte periodische Syndrome CAPS (Muckle Wells Syndrom, Familiäre Kälteurtikaria (FCAS), CINCA (Chronisch-infantile neuro-kutaneo-artikuläre)/NOMID- (Neonatal-Onset-Multisystem Disease )
Hierbei handelt es sich um eine Gruppe von Erkrankungen, bei denen eine (jeweils andere) Mutation im NLRP3 (NLR family, pyrin domain containing 3) Gen vorliegt. Während das Alter bei Symptombeginn bei CINCA/NOMID bei unter einem Jahr liegt, können sich die Symptome beim Muckle-Wells Syndrom auch bis zum 20. Lebensjahr oder sogar später eindeutig zeigen.
Allen diesen Krankheitsbildern ist eine generelle Erschöpfbarkeit und ein quaddelförmiger Hautausschlag im Schub sowie eine Bindehautentzündung bzw. bei CINCA/NOMID eine Panuveitis (Augenentzündung) gemein. Die Besonderheit bei der familiären Kälteurtikaria ist eine Auslösung von Schüben durch Kälte. Beim MWS besteht eine auffällige Innenohrschwerhörigkeit, ähnlich in Kombination mit einer aseptischen Meningitis auch bei NOMID.
Die Dauer der Schübe liegt bei 1 - 2 Tagen (bei NOMID gibt es keine Angaben zur Schubdauer).
Tumornekrosefaktor Rezeptor assoziiertes periodisches Syndrom (TRAPS)
Auch das TRAPS kann sich im Erwachsenenalter manifestieren, meist vor dem 20. Lebensjahr. Hier dauern die Schübe deutlich länger als bei den vorgenannten Erkrankungen und äußern sich meist in Form von Myalgien, Oligoarthritiden, Exanthemen, die morphologisch sehr unterschiedlich aussehen können, oft aber wie eine Pannikulitis imponieren, einer Konjunktivitis (Bindehautentzündung) und häufig, aber nicht immer, Fieber. Die Schubdauer liegt bei ca. 14 Tagen. Hier liegt die Mutation im TNFRSF1A (Tumornekrosefaktor-Rezeptor) Gen, therapeutisch kommen zur Schubvermeidung Etanercept, der lösliche Tumornekrosfeaktor-Rezeptor (Fusionsprotein), oder der IL-1-Rezeptorantagonist Anakinra in Frage.
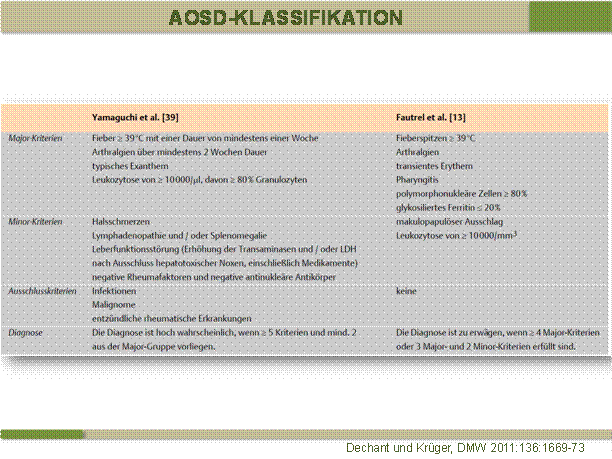
Morbus Still des Erwachsenen (Adult Onset Still`s Disease, AOSD)
Diese Erkrankung gibt es in ähnlicher Form auch im Kindesalter, bei Kindern ist dies die systemische Form der juvenilen idiopathischen Arthritis (JIA). Beim Erwachsenen ist sie, ähnlich wie die vorgenannten monogenen autoinflammatorischen Erkrankungen relativ selten. Sie äußert sich mit Fieberschüben bis 40 Grad Celsius, Arthritiden und einem im Fieberschub auftretenden, flüchtigen lachsfarbenen Exanthem. Im Labor findet sich meist eine Leukozytose und eine deutliche LDH und Ferritinerhöhung.
{kind=link}
Andere systemische Autoimmunerkrankungen
IgG4-assoziierte Erkrankung, Hyper-IgG4 Syndrom
Die igG4-assoziierte Erkrankung ist ein facettenreiches Krankheitsbild, dessen Entstehung noch nicht geklärt ist. Allen Manifestationsformen gemeinsam ist eine zunächst auf einzelne Körperregionen begrenzt erscheinende Entzündung mit konsekutiver Fibrosierung und häufig begleitender Lymphadenopathie.
Folgende Manifestationsformen bzw. Organmanifestationen sind bekannt:
{kind=link}
Andere systemische Autoimmunerkrankungen: Sarkoidose
Bei der Sarkoidose handelt es sich ebenfalls um eine entzündliche Systemerkrankung letztlich noch unklarer Pathogenese (Krankheitsentstehung). Es wird vermutet, dass es bei entsprechender genetischer Veranlagung nach infektiösem Auslöser zu einer überschießenden Reaktion (Epitheloidzellgranulome) kommt. Letztlich kann jedes Organ von einer Sarkoidose betroffen sein, allerdings sind die Häufigkeiten der Organmanifestationen unterschiedlich:
Organmanifestationen der Sarkoidose
{kind=link}
Bei der pulmonalen Sarkoidose werden verschiedene radiologische Stadien unterschieden:
- Stadium 0: unauffälliger Thorax bei extrapulmonalem Befall
- Stadium 1: bihiläre Lymphadenopathie ohne sichtbare Lungenbeteiligung, Hilusvergrößerung in der Regel beidseits
- Stadium 2: bihiläre Lymphadenopathie mit Lungenbeteiligung, die Lunge zeigt eine retikulonoduläre Zeichnung
- Stadium 3: Lungenbefall ohne sichtbare Lymphadenopathie
- Stadium 4: Lungenfibrose mit Funktionsverlust der Lunge
Eine Sonderform ist das Löfgren Syndrom mit einem entzündlichen Ödem unter der Haut eines oder beider Unterschenkel und Knöchel, manchmal auch Arthritis und/oder Tenosynovitis der oberen Sprunggelenke, einem Erythema nodosum in diesem Bereich und einer bihilären Lymphadenopathie (beidseitiger Befall der Lymphknoten in der Lunge).
Sprechen Sie uns an

Inke Jonschker
Sekretariat
- Nachricht schreiben
- (040) 18 18 81 - 11 24
- (040) 18 18 81 - 48 00